Intelligent Computational Genomics
发布时间:2020-12-08 发布人: 浏览次数:
Intelligent Computational Genomics
A key goal in biology is understanding the genetic basis for phenotypic diversity. High-throughput sequencing increasingly enables the detection of genetic differences between individuals. Nevertheless, identifying causal relationships between genotypic and phenotypic variation remains a key focus for evolutionary biology, human genetics, and plant breeding. We are interested in developing and applying new statistical models and algorithms to genome-wide inter- and intraspecific genome comparisons in order to investigate the causal genes/loci underlying trait diversity.
Gap-free chromosome-scale genome assembly
High-quality genome assembly has wide applications in genetics and medical studies. However, achieving gap-free chromosome-scale assemblies using high-throughput sequencing can still be a challenge. We implemented GALA (Gap-free long-read assembler https://github.com/ganlab/gala), a statistical framework for genome assembly through a multi-layer computer graph that identifies mis-assemblies within preliminary assemblies or chimeric raw reads and partitions the data into chromosome-scale linkage groups. The subsequent independent assembly of each linkage group generates a gap-free assembly free from the mis-assembly errors that usually impede existing workflows. This flexible framework also allows us to integrate data from various technologies such as Hi-C, genetic maps, and even motif analyses forde novoor reference-guided assemblies. GALA is also the first practical computational framework on chromosome-by-chromosome assembly.
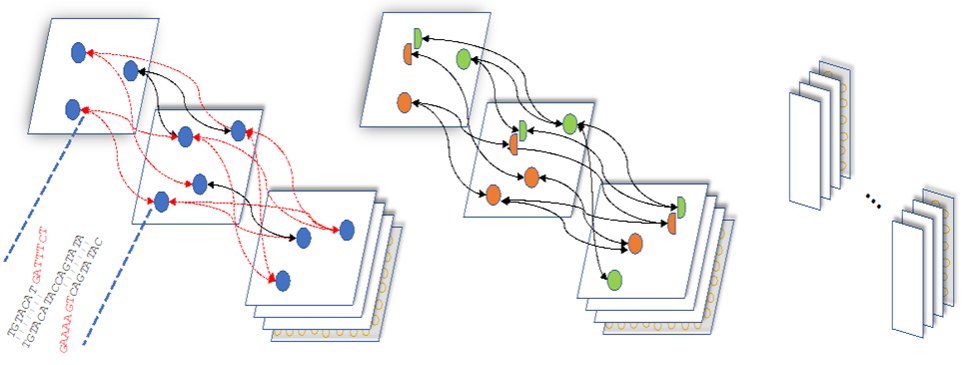
Figure 1. GALA implements a statistical framework for chromosome-by-chromosome assembly. The preliminary assemblies and raw reads are aligned against each other and encoded into a multi-layer graph. The conflicting alignments, indicated in red, are then removed iteratively by splitting the nodes involved. New edges are assigned accordingly. Nodes are then clustered into different linkage groups for independent assembly.
Structural variation studies
Many structural variants are associated with trait diversity. However, the genome-wide study is still limited because of computational challenges in their reliable identification. The long-term commitment of the group to genome assembly provides us a competitive edge in this field. For example, IMR/DENOM developed in the group can reliably detect insertions, deletions (INDEL), and structural variants (SV) such as those two variants in FRI, which are regarded as challenging to most algorithms. This allows us to perform genome-wide analysis in order to investigate the role of INDELs and SVs. Our study showed that a multi-allelic artefact caused by inconsistent alignments was a key obstacle for testing the association of insertion and deletion polymorphisms (INDELs) as well as for integrated association methods such as burden testing. To address this problem, we developed the Irisas software. This synchronises variants and integrates the impact of SNPs, INDELs, and structural variants for burden testing. We also identified novel trait loci that previous SNP-based association studies failed to map and which contain established candidate genes. We are also applying the method to explore the influence of structural variation on the genetic architecture of trait diversity.
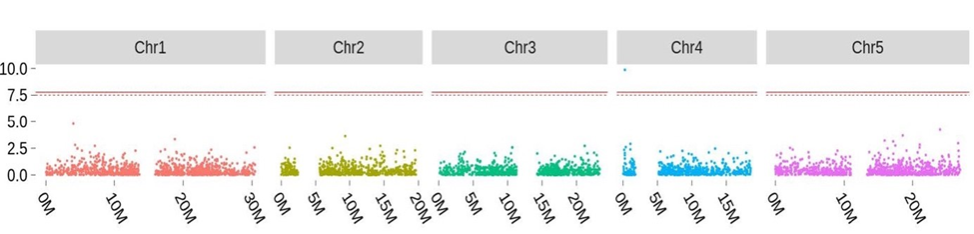
Figure 2: Integrating the effect of SNPs, INDELs, and SVs for burden analysis leads to the identification of a novel association locus around FRI for phenotypes related to flowering time.
Phenomics and digital crops
The group develop the 3D target reconstruction and segmentation platform for 3D models of rice plants under dense planting in the paddy field. We are interested in developing new techniques to identify computer vision-based features and their underlying genetic mechanisms for crop breeding.